EUKulele Quick Start¶
We recommend consulting the full documentation to explore all of the EUKulele features, capabilities, and intended usage. A quick start approach for annotating eukaryotic metagenome-assembled genomes and metatranscriptomes are outlined below using EUKulele-provided databases and default parameters.
Installation¶
The conda installation is the most straightforward approach and the environment will contain all dependencies needed to run EUKulele. If you do not already have conda installed on your machine, see the conda installation documentation here. Other installation options are included under Installation and Invocation:
conda create -n EUKulele
conda activate EUKulele
conda install -c akrinos -c bioconda -c conda-forge EUKulele
Generalized flow of EUKulele annotation¶
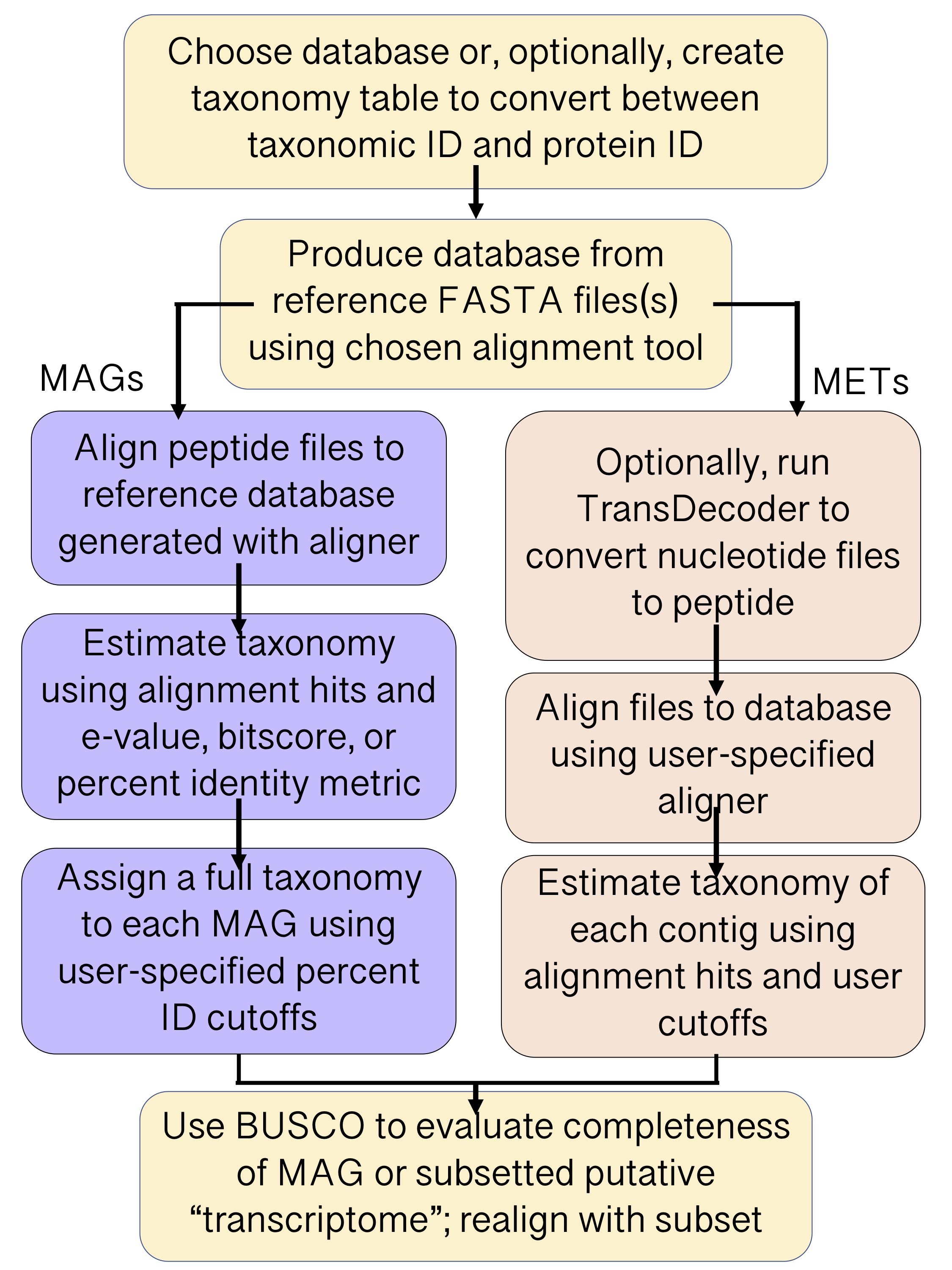
Metagenome-assembled genome (MAG) annotation¶
EUKulele can determine the taxonomic identity of binned MAGs using their consensus contig annotations. It run using the following command:
EUKulele --sample_dir/output_directory -m mags
where output_directory contains one or more assembly fasta files with the extension .faa (it is recommended that MAG files are provided in protein format; see Using EUKulele). See Parameters for other file extension accommodations. This will annotate the assemblies using the MMETSP database (default) and DIAMOND aligner (default).
Useful output is located in:
output/max-level-mag - the majority level annotation by level (supergroup - species) for each mag and the proportion of proteins that share that same annotation
output/levels_mags - consensus annotations based on the majority of annotated contigs at each classification level
output/core_taxonomy_estimation - contains contig annotations for core (shared) genes among MAGs as determined by BUSCO. Useful for identifying core genes among strains/variants during pangenomic analyses
output/busco_assessment - BUSCO assessment for the most abundant taxonomic annotation at each classification level. Provides information on estimated MAG completion based on conserved eukaryotic genes expected to be present in a full genome.
Metatranscriptomic (MET) annotation¶
EUKulele can be run on metatranscriptomic assemblies using the following command:
EUKulele --sample_dir /output_directory -m mets
where output_directory contains one or more assembly fasta files with the extension .fna. See Parameters for other file extension accommodations. This will annotate the input assemblies using the MMETSP database (default) and DIAMOND aligner (default).
Useful output files will be located in:
output/taxonomy_estimation - contains each contig’s annotation and the least common ancestor (LCA) classification threshold determined
output/taxonomy_visualization - shows the breakdown of contig annotations at the seven classification levels (Supergroup, Division, Class, Order, Family, Genus, Species) for the mixed community.
If you have any issues using EUKulele or suggestions to improve the software, please submit an issue on GitHub.